Behind the Paper: Enabling Multi-Objective Antibody Optimization
This PhD student blog post explores the use of machine learning for simultaneously optimizing antibody affinity and specificity, which could help accelerate drug development.
This PhD student blog post explores the use of machine learning for simultaneously optimizing antibody affinity and specificity, which could help accelerate drug development.
First published online at the Nature Portfolio Bioengineering Community.
View the original post here.
Challenges in Antibody Optimization
Antibody therapeutics have emerged as one of the most promising therapeutic modalities, transforming the treatment of many diseases. However, their development remains extremely challenging. While their success is a result of their combination of drug-like biophysical properties, it remains very difficult to identify candidate molecules with optimal combinations of biophysical properties. This is due in part to the fact that these properties exhibit strong tradeoffs and making improvements to one property can be detrimental to others. Furthermore, antibody design space is massive, and therapeutic antibodies are rare within that space, which has necessitated the development of new technologies for their development.
One technology that has emerged as a workhorse for therapeutic antibody development is directed evolution, the cyclic process of antibody sequence diversification and subsequent selection. The most efficient use of directed evolution comes in the form of library production and screening, during which thousands of different antibody sequence variants are generated and selected based on specific molecular properties. In concert with deep sequencing technology, library screening facilitates investigation of large swaths of antibody sequence space in search of these rare therapeutic antibodies.
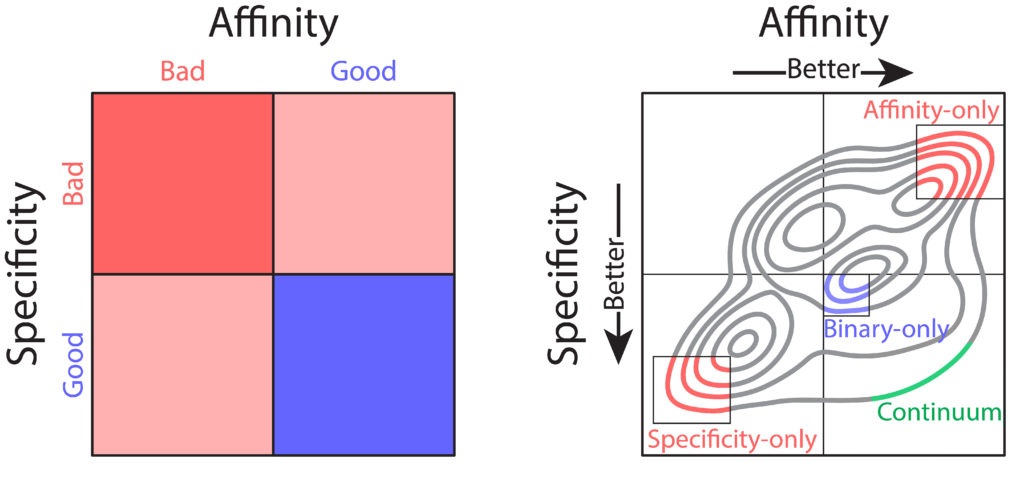
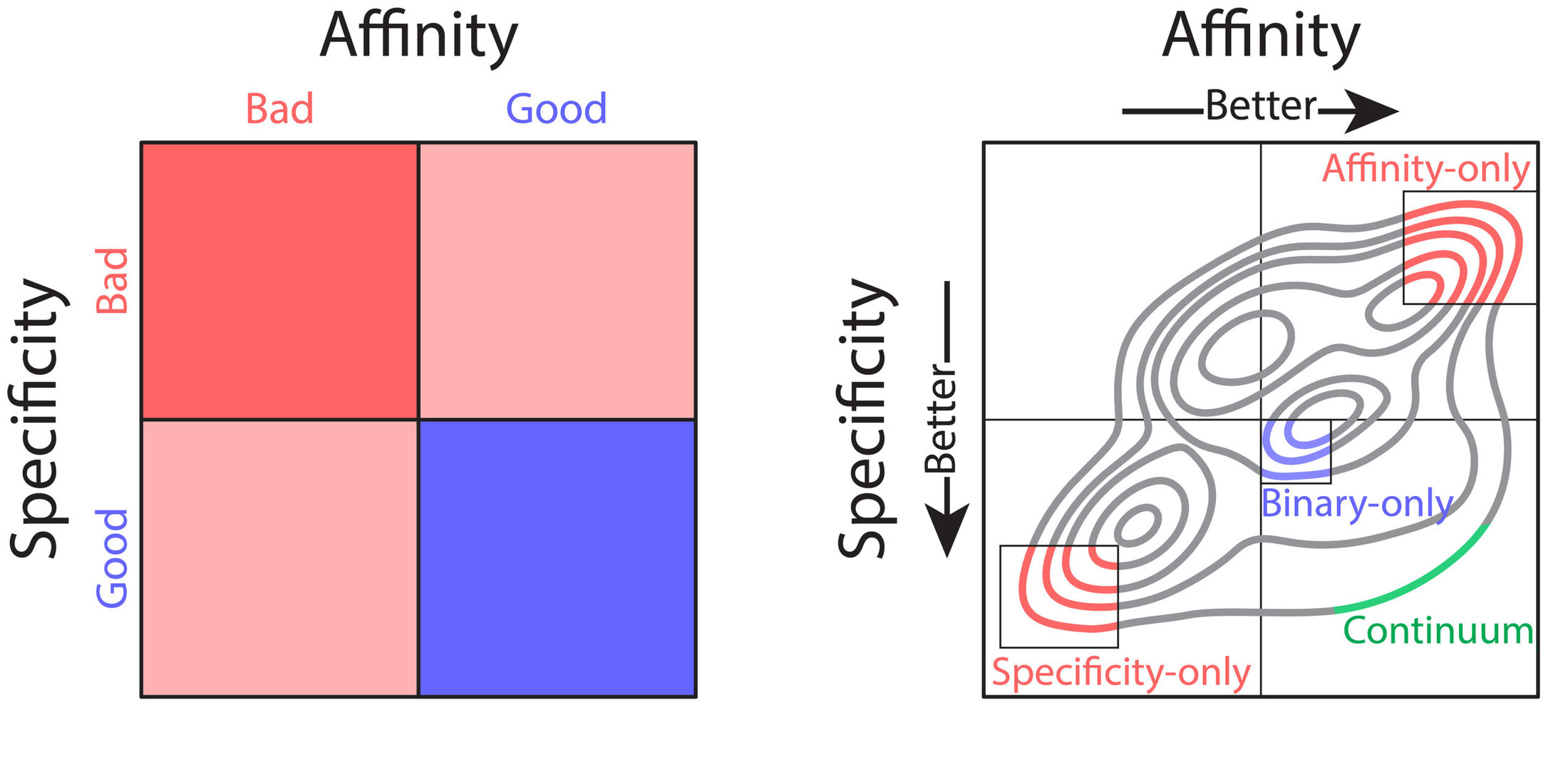
Nevertheless, these technologies have several shortcomings that limit their practical use for therapeutic antibody development. Most notably, library screening and deep sequencing provide coarse-grained information about antibody sequence behavior, typically in the form of binary labels (i.e., binding vs non-binding), instead of providing continuum values for affinity. This complicates the development process, as optimization of properties such as affinity requires fine-grained information (i.e., high binding vs very high binding). In combination with the strict tradeoffs between properties, these shortcomings impede development of drug-like antibodies. What would be ideal is prediction of continuum values for properties that could be used in a multi-objective, or Pareto, optimization, to achieve simultaneous optimization of antibody affinity and properties such as specificity (non-specific binding). Pareto optimization is particularly powerful because it enables identification of the set of variants that are maximally optimized, along what is called the Pareto frontier. At this frontier no further improvements can be achieved to one property without sacrificing other properties. To this end, we sought to transform the coarse-grained information gained from library screening and deep sequencing into usable, continuous metrics for property evaluation through the application of machine learning.
Enabling multi-objective antibody optimization
We sought to optimize a therapeutic antibody, emibetuzumab, which has high on-target binding (affinity) but suffers from high off-target binding (non-specific binding), limiting its clinical potential. We wanted to co-optimize the antibody, hoping to achieve substantial reduction of non-specific binding while maintaining or increasing its affinity. We first generated a library of millions of sequence variants, screening it for both affinity and non-specific binding. Deep sequencing was used to generate large datasets of sequences coarsely labeled as “high” or “low” for both affinity and non-specific binding. With the application of various machine learning models, it was easy to achieve high classification accuracies, upwards of 90%. But despite these high accuracies, classification was of limited use for optimization. Due to large intraclass variability, antibody variants classified as optimal for both affinity and non-specific binding often exhibited combinations of these properties that were not therapeutically acceptable. Most often, these variants exhibited desirable high specificity but only modest affinity that would be too weak for clinical use.
One particular classification algorithm we used was linear discriminant analysis (LDA). LDA works by first projecting the high-dimensional feature vectors into a one-dimensional space, then finding the optimal classification threshold along that one-dimensional projection. Using LDA, we were able to achieve high classification accuracies (93% for both affinity and non-specific binding). Interestingly, inspection of the LDA projections revealed a range of predicted values, with particularly notable variation within property classes (i.e., affinity or specificity). We hypothesized that this variation may reflect the intraclass variation of continuous property measurements. To test this hypothesis, we employed time-consuming low-throughput methodology to acquire continuous experimental measurements of affinity and non-specific binding for a large set of emibetuzumab mutants. Surprisingly, we found that the LDA projections, acquired solely from sequences with binary labels, correlated strongly with continuous measurements, providing us with a proxy for continuous affinity and non-specific binding measurements for every mutant in our library. By identifying continuum values at a large scale, we could pursue multi-objective optimization of both affinity and non-specific binding, through what is known as a Pareto optimization.
In our Pareto optimization, we plotted the continuum predictions of both affinity and non-specific binding for all sequences in the library against one another. This type of visualization facilitates simple identification of variants with predictions of improved behavior along the Pareto frontier. The plot also revealed a remarkably strong tradeoff between affinity and non-specific binding, in alignment with our experimental experience. As expected, our starting antibody, emibetuzumab, was located in the region of the Pareto plot that corresponded to both high affinity and high non-specific binding, but was not at the Pareto frontier. This suggested that emibetuzumab could be co-optimized for both properties as sequences with predictions of higher affinity and lower non-specific binding could be identified.
Towards co-optimizing emibetuzumab, we used our novel computational models to predict additional sequences with co-optimal properties. Impressively, over one third had co-optimized properties and a majority had therapeutically relevant property combinations. Our best variant had 30% higher binding affinity and 70% lower non-specific binding and boasted several other drug-like properties.
Beyond ‘affinity-first’ antibody design
Our work demonstrates that library screening and deep sequencing can be combined with machine learning to predict continuous metrics that are strongly correlated with affinity and non-specific binding at a large scale, and that these continuous predictions can be used to optimize multiple antibody properties simultaneously. Current antibody development techniques generally focus on optimizing affinity first, and then triaging other properties that are found to be suboptimal in later stages. Our work has several key implications for future antibody optimization to move beyond the “affinity-first” paradigm.
First, it emphasizes the importance of multi-objective, rather than affinity-first, antibody optimization. Our work builds on several other papers that demonstrate that antibody biophysical properties are oftentimes linked, illustrating that optimizing one property at a time has widespread effects on other properties. The quantification and visualization of the tradeoff between affinity and non-specific binding for our emibetuzumab library further supports this idea. Another aspect of multi-objective optimization is the possibility of tailoring antibody properties to specific biological contexts with unique therapeutic optimums, as opposed to simply maximizing target affinity.
Our work also stands as proof of concept with countless future directions for exploration. We focused on a simple linear model and a small set of amino acid sequence features. Both components present opportunities for further customization and optimization. Increasingly complex datasets can be analyzed with increasingly complex models and increasingly descriptive feature embeddings to improve efficiency of sequence-space exploration and the accuracy of predictions. Overall, this suggests that we may be able to utilize larger antibody libraries for directed evolution of therapeutic antibody development as accurate information can be better extracted from less data.
In summary, our work provides a novel way to identify continuous measures of affinity and specificity from sorted antibody libraries. Multi-objective antibody optimization may help improve antibody design and accelerate therapeutic antibody approvals, ultimately improving patient health.



